
Venkatesh Lab
E. coli metabolism
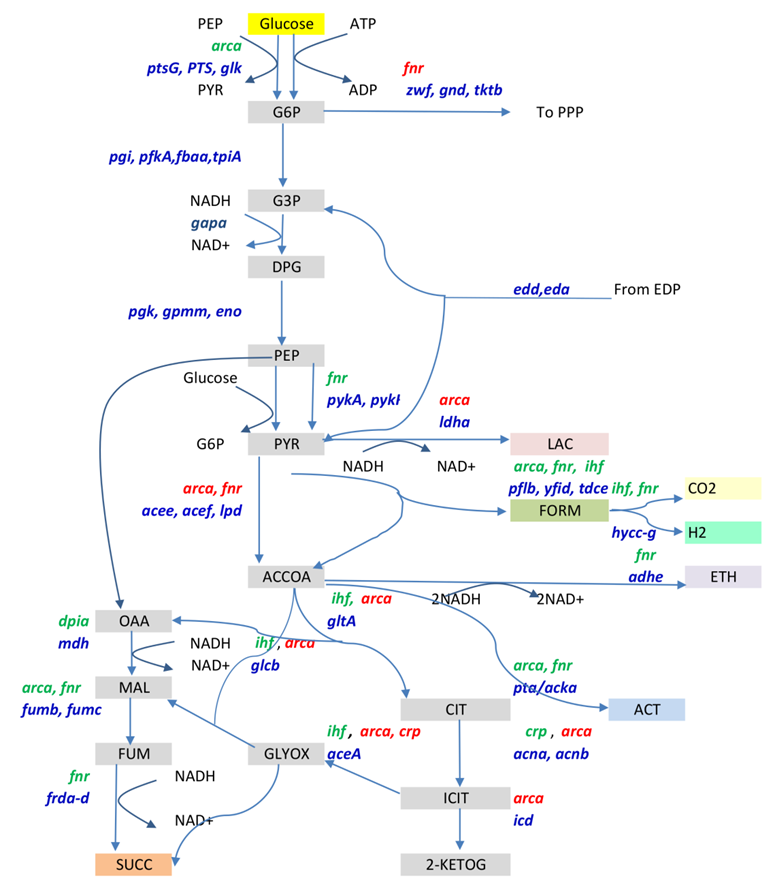 E. coli is a facultative anaerobic bacterium capable of surviving with (aerobic) or without (anaerobic) oxygen. We study the effect of key transcriptional regulators on the growth of E. coli under different aeration conditions. Transcriptional regulators or factors (TFs) play a key role in regulating the flux distribution in vivo. Many studies enumerate the role of important global transcription factors that regulate metabolism under various conditions. However, these studies employ single deletion mutants of these TFs. It is now clear that due to cross interactions with several genes, a single deletion mutant may not enumerate the specific roles of a given TF. Thus it is important to study the impact of perturbation due to the deletion of key transcriptional regulators on metabolic network in E. coli comprehensively, under different conditions in comparison with the wild type strain.
E. coli is a facultative anaerobic bacterium capable of surviving with (aerobic) or without (anaerobic) oxygen. We study the effect of key transcriptional regulators on the growth of E. coli under different aeration conditions. Transcriptional regulators or factors (TFs) play a key role in regulating the flux distribution in vivo. Many studies enumerate the role of important global transcription factors that regulate metabolism under various conditions. However, these studies employ single deletion mutants of these TFs. It is now clear that due to cross interactions with several genes, a single deletion mutant may not enumerate the specific roles of a given TF. Thus it is important to study the impact of perturbation due to the deletion of key transcriptional regulators on metabolic network in E. coli comprehensively, under different conditions in comparison with the wild type strain.
In metabolism, the substrate intake is directed towards three main objectives, namely, energy balance (ATP synthesis and consumption), biomass synthesis (growth and biopolymer synthesis) and redox balance (NADH synthesis and consumption). The resulting growth rate of an organism is an optimized function of the three competing objectives. Here, we ask the relevant question as to whether the global transcriptional regulators have a role in optimizing growth to achieve the three objectives. The exact mechanism of how the growth phenotype is determined via the optimization of energy requirements, redox balance and biosynthesis through transcriptional regulation via combinatorial mutants has not been addressed before. We study the effect of deletion of key transcription regulators and combinations of them on the above mentioned objectives and rationalize the growth observed based on them.
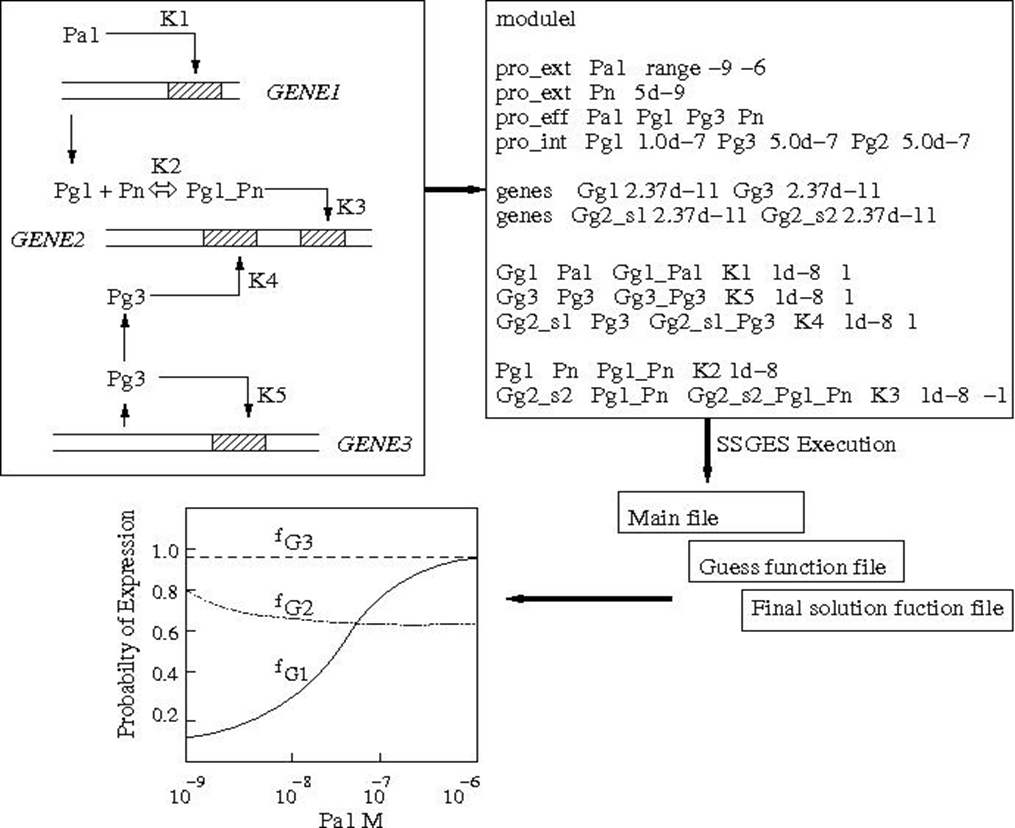 Network modeling helps in rationalizing the interactions between various components linking genotype to phenotype. Studies involving combinatorial deletion mutants would result in quantitative measurements at the level of gene and protein expression and the resulting reaction rates (fluxes) in the metabolic network. We have developed a steady state modeling formalism for gene expression (SSGES) which includes molecular mechanisms that help in simulating microarray data with high accuracy and can be used to study the effect of both structural and parametric perturbation on gene expression. SSGES includes mechanistic details such as stoichiometry, protein–DNA and protein–protein interactions, translocation of regulatory proteins and auto-regulation. A similar steady state signaling network model will be developed connecting the inputs from the environmental cues to the output of TF expression. The expression of TF will be connected to the metabolic network. Metabolic network consists of a complex set of highly interconnected enzyme catalyzed reactions. A particular phenotype is characterized by a set of such reactions catalyzed by various enzymes present in its genomic capability. We use flux balance analysis which require only the stoichiometric information of the metabolic network. Our intention is to combine steady state models for the three subcomponents and predict the growth rate of the organism under transcription factor deletion and environmental stress. Such a multi-scale model will be used to rationalize the data obtained at the multiple levels through experiments. We also perform long term evolutionary experiments by selecting the fittest growing colony under a specific condition in every generation of low growth combinatorial mutants to assess if selection results in any fitness advantage and if so what mutations lead to such an advantage by genotypic and phenotypic characterization of the evolved mutant.
Network modeling helps in rationalizing the interactions between various components linking genotype to phenotype. Studies involving combinatorial deletion mutants would result in quantitative measurements at the level of gene and protein expression and the resulting reaction rates (fluxes) in the metabolic network. We have developed a steady state modeling formalism for gene expression (SSGES) which includes molecular mechanisms that help in simulating microarray data with high accuracy and can be used to study the effect of both structural and parametric perturbation on gene expression. SSGES includes mechanistic details such as stoichiometry, protein–DNA and protein–protein interactions, translocation of regulatory proteins and auto-regulation. A similar steady state signaling network model will be developed connecting the inputs from the environmental cues to the output of TF expression. The expression of TF will be connected to the metabolic network. Metabolic network consists of a complex set of highly interconnected enzyme catalyzed reactions. A particular phenotype is characterized by a set of such reactions catalyzed by various enzymes present in its genomic capability. We use flux balance analysis which require only the stoichiometric information of the metabolic network. Our intention is to combine steady state models for the three subcomponents and predict the growth rate of the organism under transcription factor deletion and environmental stress. Such a multi-scale model will be used to rationalize the data obtained at the multiple levels through experiments. We also perform long term evolutionary experiments by selecting the fittest growing colony under a specific condition in every generation of low growth combinatorial mutants to assess if selection results in any fitness advantage and if so what mutations lead to such an advantage by genotypic and phenotypic characterization of the evolved mutant.