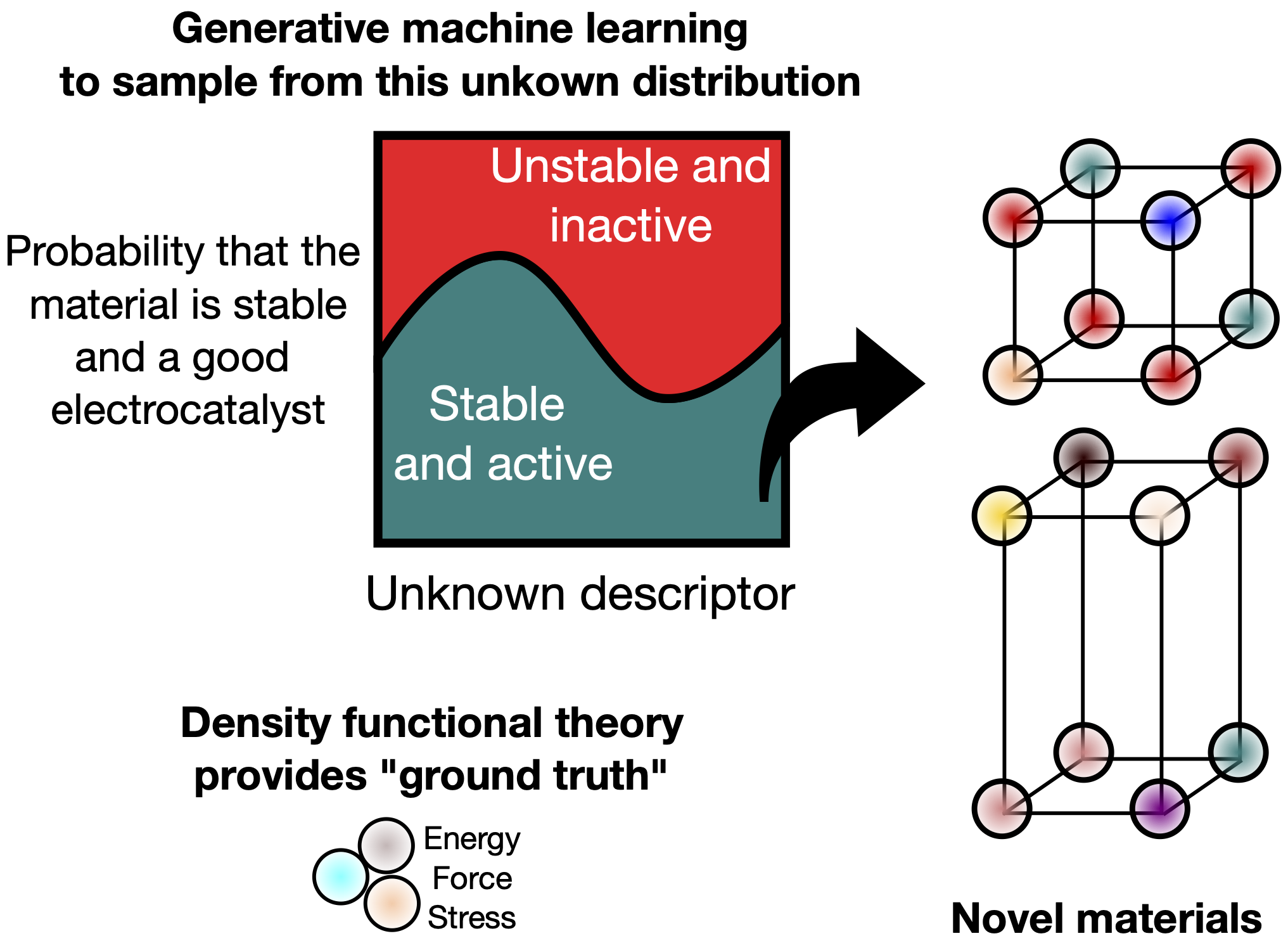
Generative Machine Learning for Designing Stable Catalysts and Electrodes for Electrochemical Hydrogen Production
The goal of this project is to discover novel and stable materials that can be used as catalysts and electrodes for electrochemical hydrogen production. These materials are essential to creating efficient water electrolyzers, which use electricity to split water into hydrogen and oxygen.
A well-known example is platinum, which is an excellent catalyst for these reactions but is also very rare and expensive. To make hydrogen production more practical and affordable, we need to find alternative materials that are both efficient and stable.
To find these materials, we use machine learning models trained on data from physics-based simulations. These simulations tell us whether certain arrangements of atoms are likely to form stable materials. These models learn from this data and can generate new, promising material candidates.
In particular, we focus on developing generative models which create entirely new atomic structures and compare them with traditional search methods. This approach can speed up the discovery of next-generation materials for hydrogen generation technologies.
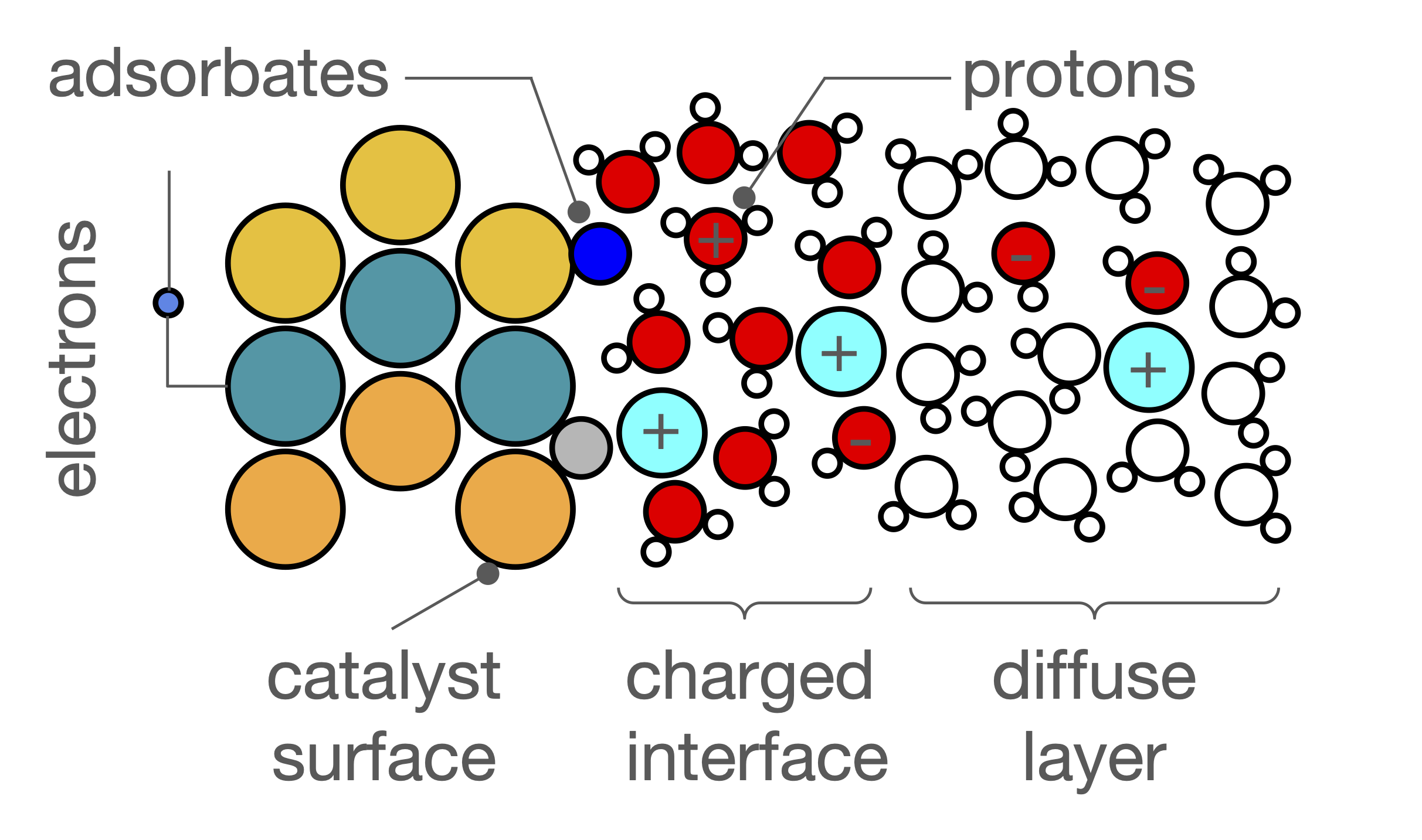
Advancing Density Functional Theory Based Methods to Understand and Design Electrocatalysts
We develop new methods based on density functional theory (DFT) to simulate chemical reactions on the surface of electrocatalysts, materials that drive reactions using electricity. These methods allow us to compute reaction rates under realistic conditions, such as under a constant electrical potential, which better reflects what happens during actual experiments. These state-of-the-art simulation techniques help us design more effective catalysts for use in electrochemical systems.
To explore how these materials behave over longer time and length scales, we also apply state-of-the-art machine learning models. These models, known as interatomic potentials, allow us to run molecular dynamics simulations that reveal how atoms move and interact during reactions. These simulations helps us understand the most active sites on catalyst surfaces and uncover design principles for improving their performance.
A key application of this work is in the alkaline hydrogen evolution reaction, where water is split in a basic environment to produce hydrogen fuel. Our goal is to help develop catalysts that are more efficient, durable, and cost-effective for clean hydrogen production.
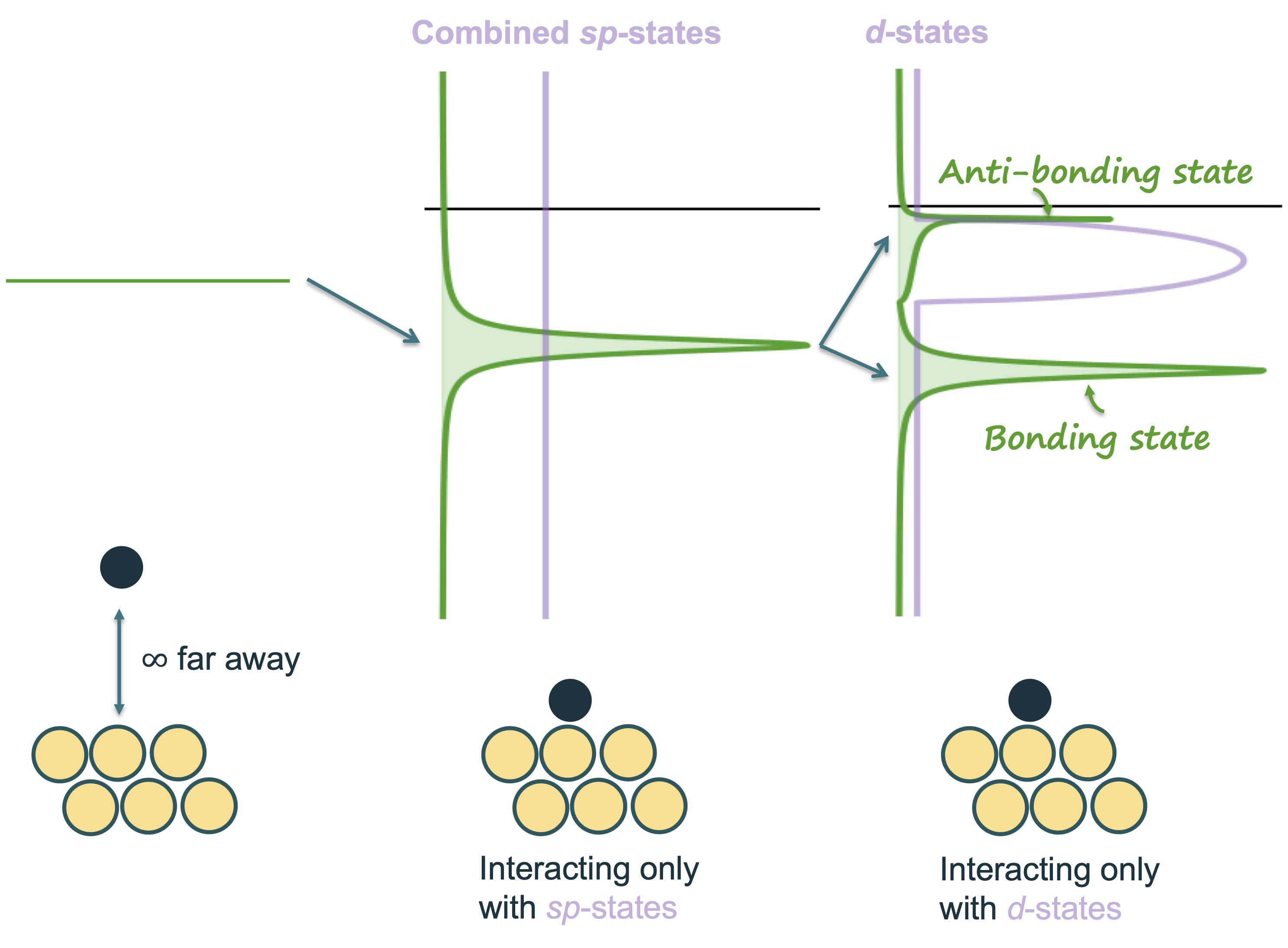
Developing General Theories of Chemisorption for Catalyst Design
This project focuses on building a deeper theoretical understanding of how and why catalysts work. We develop theoretical insights as to how small changes to a catalyst’s surface can affect its performance. In particular, we study how the electronic structure of a catalyst surface (like the position of its d-band) controls how strongly molecules bind to it. This binding strength determines whether a catalyst is highly reactive, unreactive, or even poisoned by certain molecules.
For example, metals such as gold and silver bind molecules weakly because their d-band lies far below the Fermi level, which makes them less reactive. On the other hand, some metals bind too strongly, which can poison the surface. By understanding and predicting these interactions using quantum mechanical simulations like density functional theory (DFT), we can explain trends in catalytic activity across many different materials.
We are now extending this theory to include more realistic conditions, such as the effect of electric fields, interactions between adsorbed molecules on the surface, and complex surfaces made of multiple elements. The goal is to develop a general, physics-based theory of how surface chemistry works, which can guide the design of better catalysts for chemical and electrochemical reactions.